Research
Data-driven robust control for insulin therapy
The artificial pancreas aims to automate treatment of type 1 diabetes (T1D) by integrating insulin pump and glucose sensor through control algorithms. However, fully closed-loop therapy is challenging since the blood glucose levels to control depend on disturbances related to the patient behavior, mainly meals and physical activity.
To handle meal and exercise uncertainties, in our work we construct data-driven models of meal and exercise behavior, and develop a robust model-predictive control (MPC) system able to reject such uncertainties, in this way eliminating the need for meal announcements by the patient. The data-driven models, called uncertainty sets, are built from data so that they cover the underlying (unknown) distribution with prescribed probabilistic guarantees. Then, our robust MPC system computes the insulin therapy that minimizes the worst-case performance with respect to these uncertainty sets, so providing a principled way to deal with uncertainty. State estimation follows a similar principle to MPC and exploits a prediction model to find the most likely state and disturbance estimate given the observations.
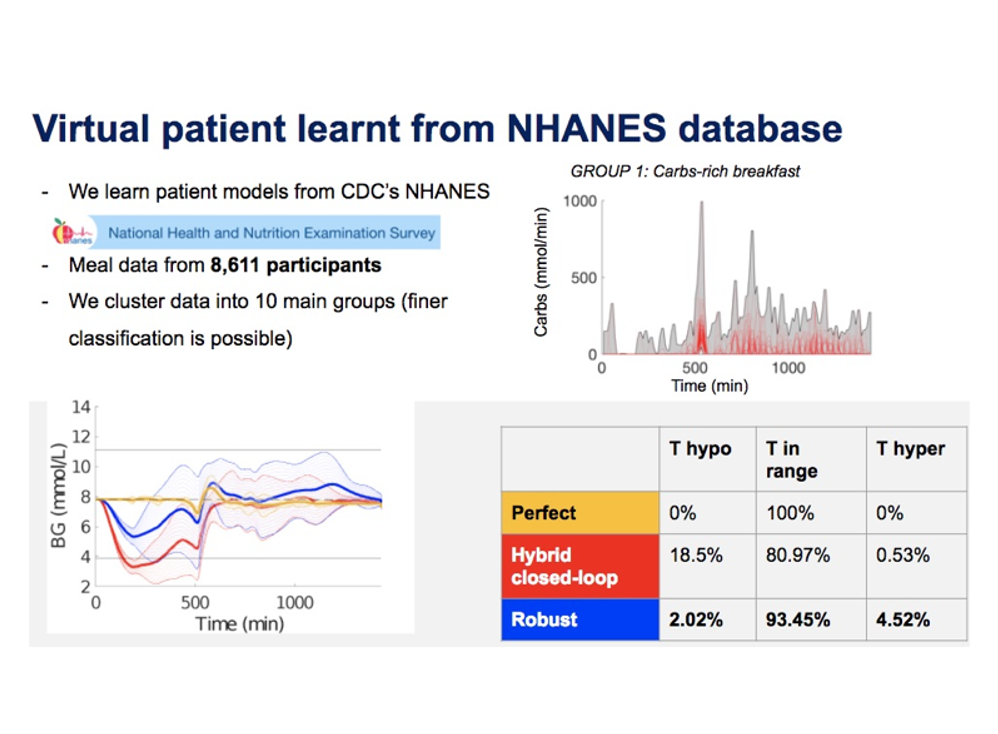
We evaluate our design on synthetic scenarios, including high-carbs intake and unexpected meal delays, and on large clusters of virtual patients learned from population-wide survey data sets (CDC NHANES).
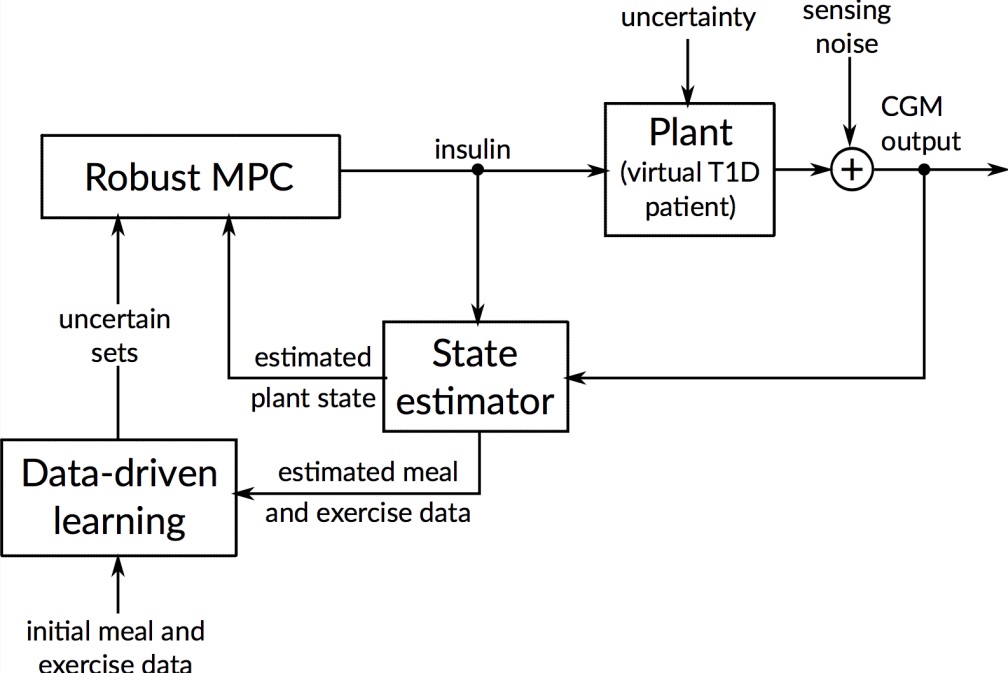
• , Data-Driven Robust Control for a Closed-Loop Artificial Pancreas. In IEEE/ACM Transactions on Computational Biology and Bioinformatics, 17 (6): 1981-1993, 2020. (BIB) (PDF) (DOI)
• , Data-Driven Robust Control for Type 1 Diabetes Under Meal and Exercise Uncertainties. In Computational Methods in Systems Biology (CMSB 2017), pages 214-232, 2017. (BIB) (PDF) (DOI)
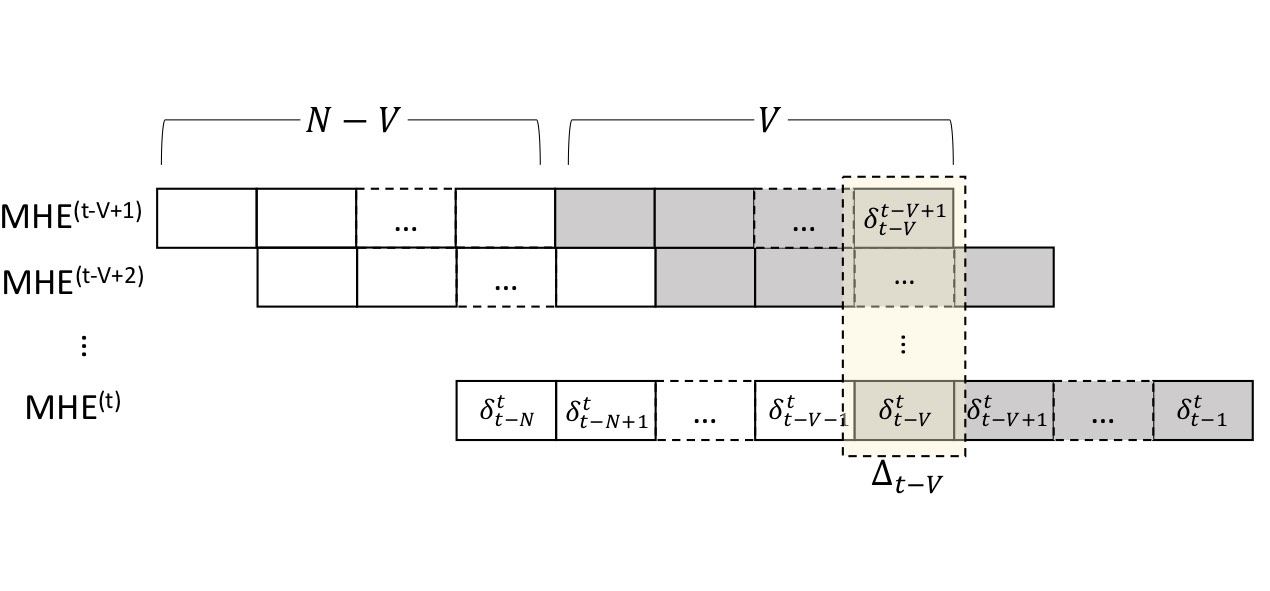
Committed Moving Horizon Estimation for Meal Detection
We introduce Committed Moving Horizon Estimation (CHME), a model-based technique for detecting and estimating unknown random disturbances in control systems. We investigate CHME in the context of meal detection and estimation method for the treatment of type 1 diabetes, where we are interested in automatically detecting the occurrence and estimate the amount of carbohydrate (CHO) intake from glucose sensor data. Meal detection and estimation is crucial in closed-loop insulin control as it can eliminate the need for manual meal announcements by the patient.
CMHE extends Moving Horizon Estimation, which alone, is not well-suited for disturbance estimation and meal detection. Indeed, accurate disturbance estimation needs both look-ahead (awareness of the disturbance effect on future system outputs) and history (past outputs to discriminate the start of the disturbance). For this purpose, CMHE aggregates the meal disturbances estimated by multiple MHE instances to balance future and past information at decision time, thus providing timely detection and accurate estimation. Applied to the detection of random meals from glucose sensor data, our method achieves an 88.5% overall detection rate and a 100% detection rate for the main meals (i.e., excluding snacks).
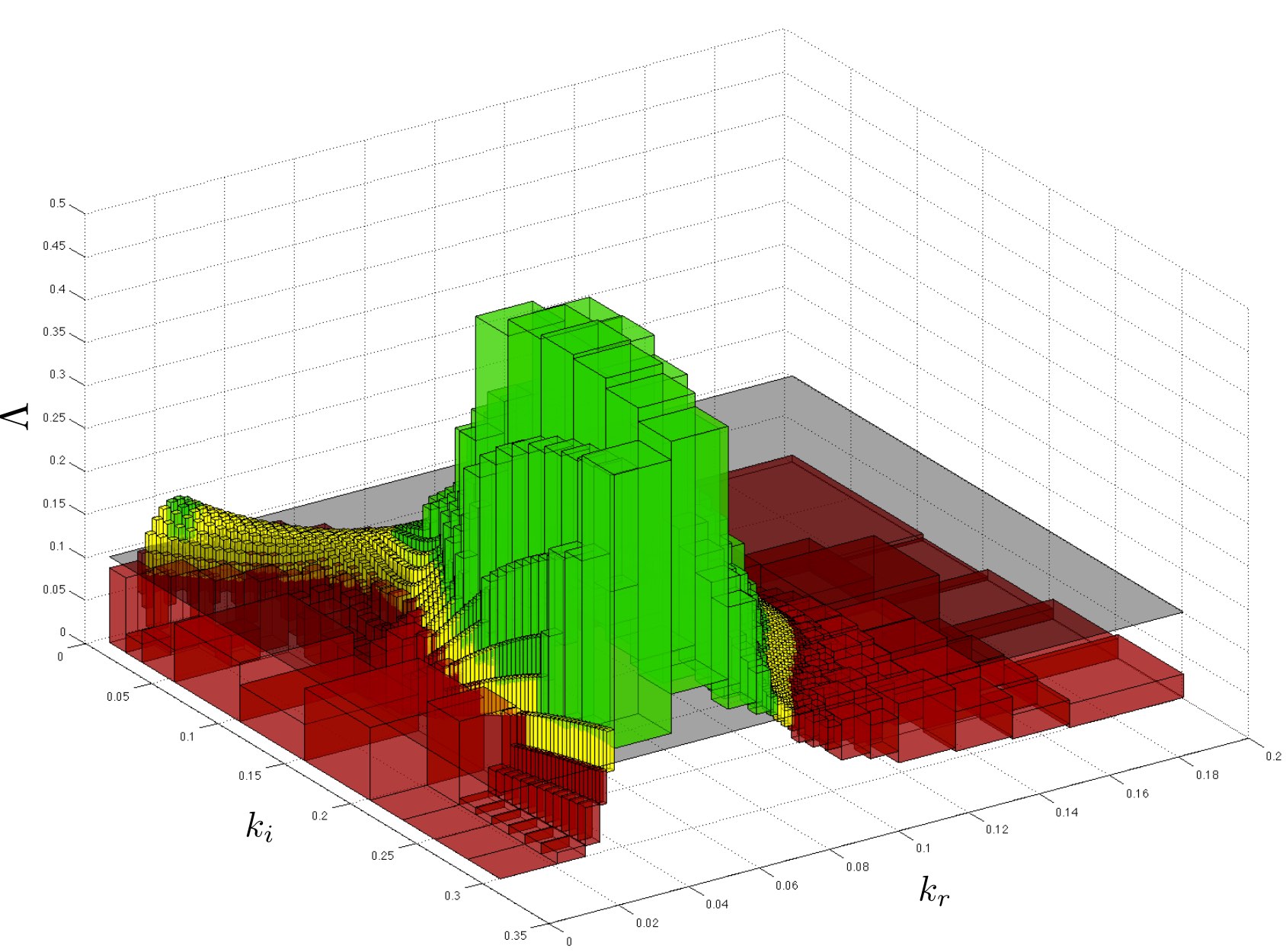
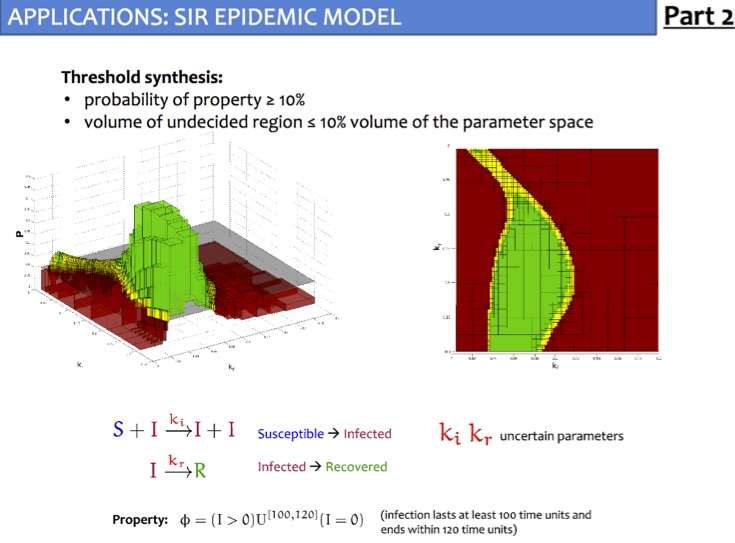
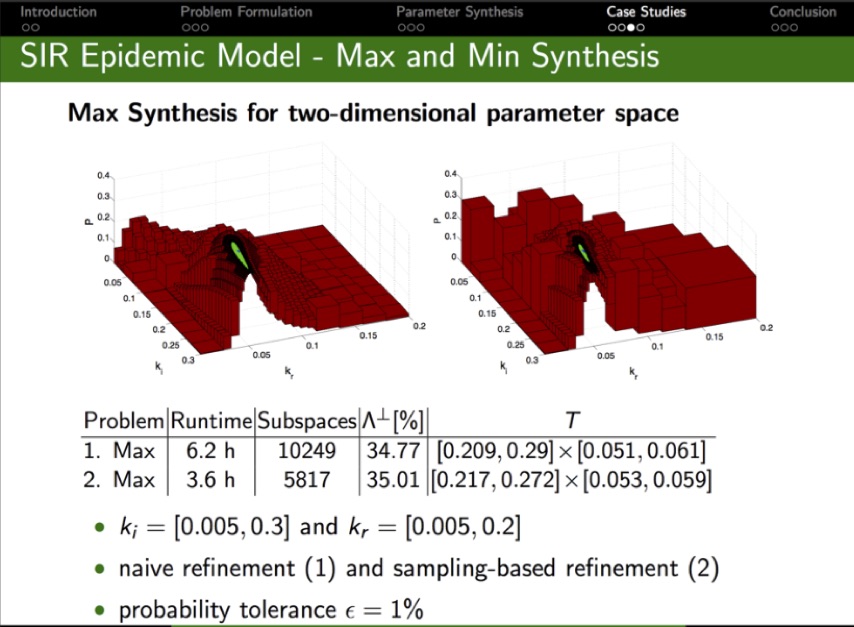
Precise parameter synthesis for continuous-time Markov chains
Given a parameteric continuous-time Markov chain (pCTMC), we aim at finding parameter values such that a CSL property is guaranteed to hold (threshold synthesis), or, in the case of quantitative properties, the probability of satisfying the property is maximised or minimised (max synthesis).
The solution of the threshold synthesis (see picture) is a decomposition of the parameter space into true and false regions that are guaranteed to, respectively, satisfy and violate the property, plus an arbitrarily small undecided region. On the other hand, in the max synthesis problem we identify an arbitrarily small region guaranteed to contain the actual maximum/minimum.
We developed synthesis methods based on a parameteric extension of probabilistic model checking to compute probability bounds, as well as refinement and sampling of the parameter space. We implemented GPU-accelerated algorithms for synthesis in the PRISM-PSY tool, which we applied to a variety of biological and engineered systems, including models of molecular walkers, mammalian cell cycle, and the Google file system.
• , Precise Parameter Synthesis for Generalised Stochastic Petri Nets with Interval Rate Parameters. In International Conference on Computer Aided Systems Theory (EUROCAST 2017), pages 38-46, 2018. (BIB) (PDF) (DOI)
• , Precise Parameter Synthesis for Stochastic Biochemical Systems. In Acta Informatica, 54 (6): 589-623, 2017. (BIB) (PDF) (DOI)
• , PRISM-PSY: precise GPU-accelerated parameter synthesis for stochastic systems. In Tools and Algorithms for the Construction and Analysis of Systems (TACAS 2016), pages 367-384, 2016. (BIB) (PDF) (DOI)
• , Precise parameter synthesis for stochastic biochemical systems. In Computational Methods in Systems Biology (CMSB 2014), pages 86-98, 2014. (BIB) (PDF) (DOI)
Optimal synthesis of stochastic chemical reaction networks
The automatic derivation of Chemical Reaction Networks (CRNs) with prescribed behavior is one of the holy grails of synthetic biology, allowing for design automation of molecular devices and in the construction of predictive biochemical models.
In this work, we provide the first method for optimal syntax-guided synthesis of stochastic CRNs that is able to derive not just rate parameters but also the structure of the network. Borrowing from the programming language community, we propose a sketching language for CRNs that allows to specify the network as a partial program, using holes and variables to capture unknown components. Under the Linear Noise Approximation of the chemical master equation, a CRN sketch has a semantics in terms of parametric Ordinary Differential Equations (ODEs).
We support rich correctness properties that describe the temporal profile of the network as constraints over mean and variance of chemical species, and their higher order derivatives. In this way, we can synthesize networks where e.g., one of the species exhibit a bell-shape profile or has variance greater than its expectation.
We synthesize CRNs that satisfies the sketch constraints and a correctness specification while minimizing a given cost function (capturing the structural complexity of the network). The optimal synthesis algorithm employs SMT solvers over reals and ODEs (iSAT) and a meta-sketching abstraction that speeds up the search through cost constraints.
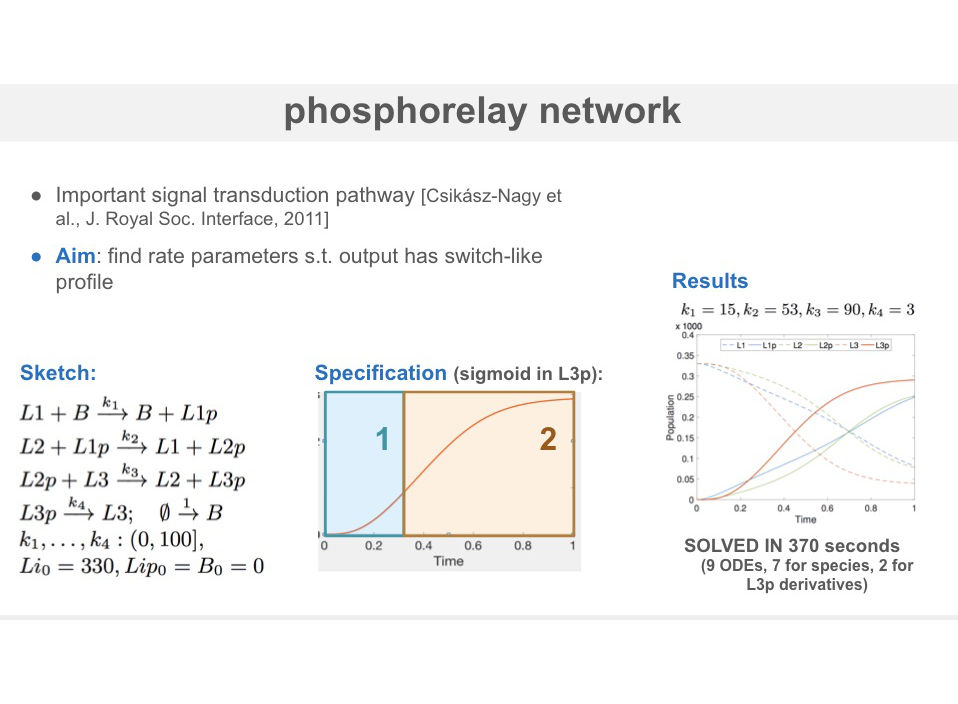
We evaluate the approach on a three key problems: synthesis of networks with a bell-shaped profile (occurring in signaling cascades), a CRN implementation of a stochastic process with prescribed levels of noise and synthesis of sigmoidal profiles in the phosphorelay network.
• , Syntax-Guided Optimal Synthesis for Chemical Reaction Networks. In International Conference on Computer Aided Verification (CAV 2017), pages 375-395, 2017. (BIB) (PDF) (DOI)
Closed-loop quantitative verification of rate-adaptive pacemakers
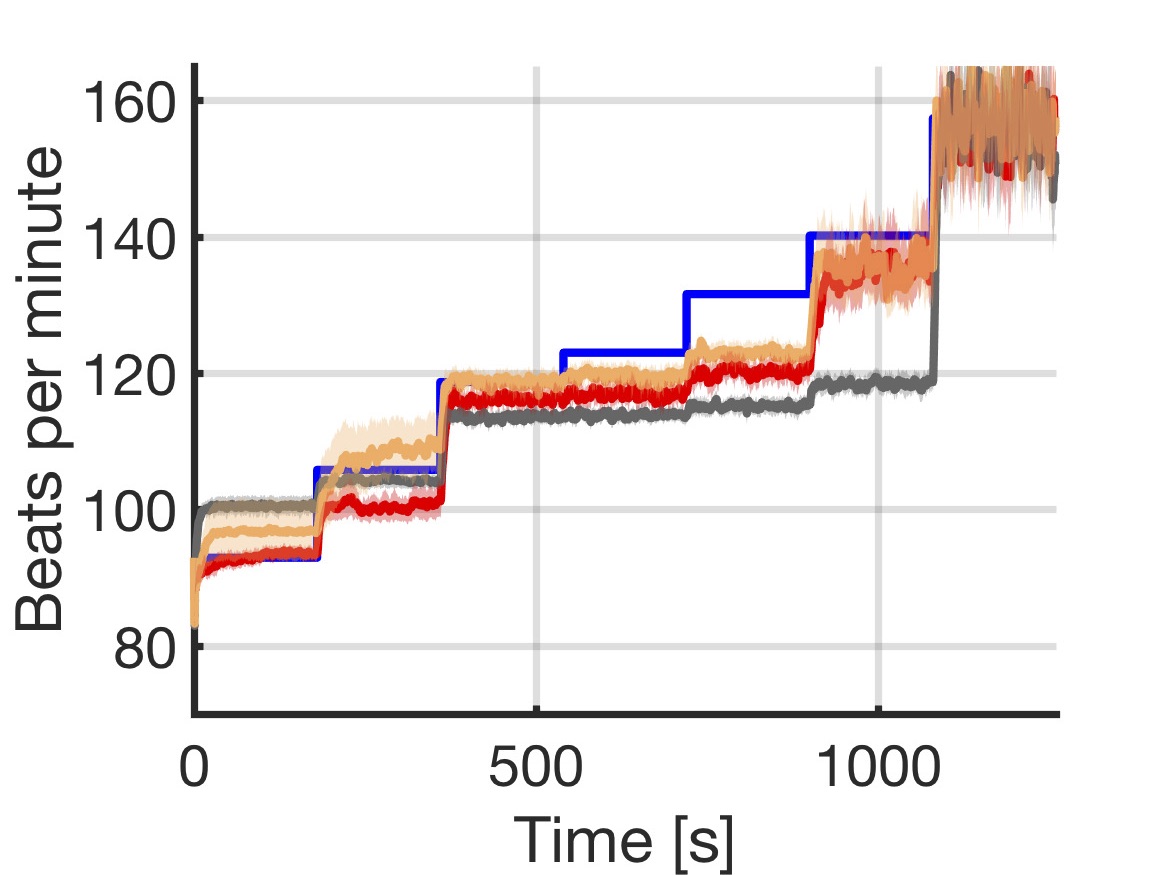
Rate-adaptive (RA) pacemakers are able to adjust the pacing rate depending on the levels of activity of the patient, detected by processing physiological signals data. RA pacemakers represent the only choice when the heart rate cannot naturally adapt to increasing demand (e.g. AV block). RA parameters depend on many patient-specific factors, and effective personalisation of such treatments can only be achieved through extensive exercise testing, which is normally intolerable for a cardiac patient.
We introduce a data-driven and model-based approach for the quantitative verification of RA pacemakers and formal analysis of personalised treatments. We developed a novel dual-sensor pacemaker model where the adaptive rate is computed by blending information from an accelerometer, and a metabolic sensor based on the QT interval (a feature of the ECG). The approach builds on the HeartVerify tool to provide statistical model checking of the probabilistic heart-pacemaker models, and supports model personalization from ECG data (see heart model page). Closed-loop analysis is achieved through the online generation of synthetic, model-based QT intervals and acceleration signals.
We further extend the model personalization method to estimate parameters from patient population, thus enabling safety verification of the device. We evaluate the approach on three subjects and a pool of virtual patients, providing rigorous, quantitative insights into the closed-loop behaviour of the device under different exercise levels and heart conditions.
• , Closed-loop quantitative verification of rate-adaptive pacemakers. In ACM Transactions on Cyber-Physical Systems, 2 (4), 2018. (BIB) (PDF) (DOI)
• , Formal modelling and validation of rate-adaptive pacemakers. In Healthcare Informatics (ICHI), 2014 IEEE International Conference on, pages 23-32, 2014. *Best Paper Award*. (BIB) (PDF) (DOI)
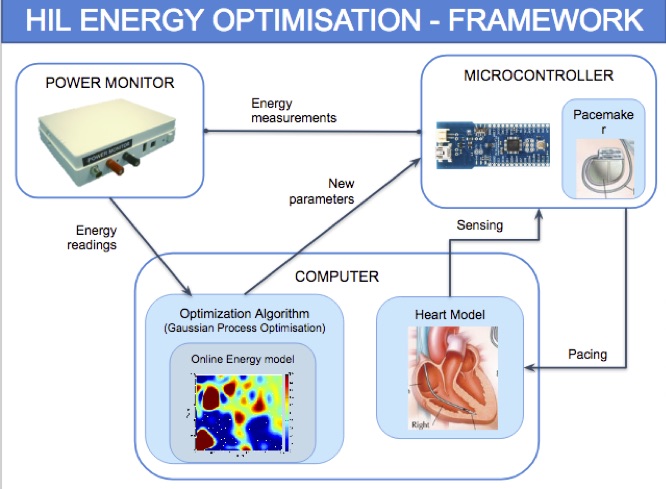
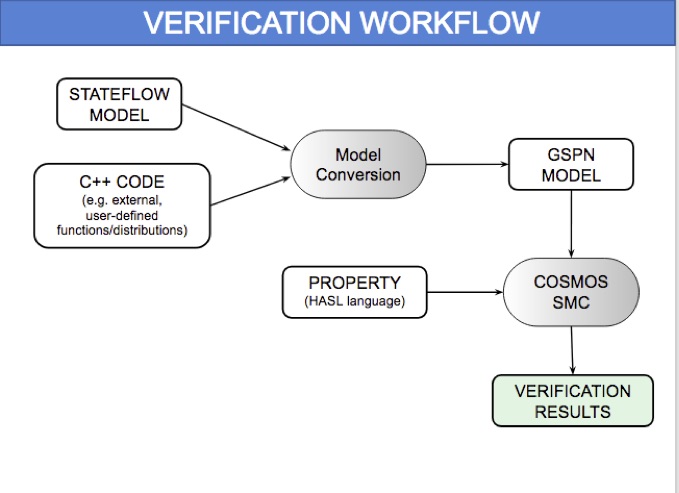
HeartVerify: Model-Based Quantitative Verification of Cardiac Pacemakers
HeartVerify is a framework for the analysis and verification of pacemaker software and personalised heart models. Models are specified in MATLAB Stateflow and are analysed using the Cosmos tool for statistical model checking, thus enabling the analysis of complex nonlinear dynamics of the heart, where precise (numerical) verification methods typically fail.
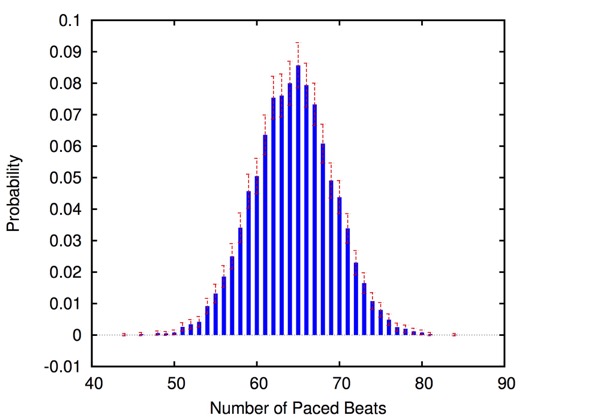
The approach is modular in that it allows configuring and testing different heart and pacemaker models in a plug-and-play fashion, without changing their communication interface or the verification engine. It supports the verification of probabilistic properties, together with additional analyses such as simulation, generation of probability density plots (see figure) and parametric analyses. HeartVerify comes with different heart and pacemaker models, including methods for model personalization from ECG data and rate-adaptive pacing.
• , Closed-loop quantitative verification of rate-adaptive pacemakers. In ACM Transactions on Cyber-Physical Systems, 2 (4), 2018. (BIB) (PDF) (DOI)
• , Estimation and verification of hybrid heart models for personalised medical and wearable devices. In Computational Methods in Systems Biology (CMSB 2015), pages 3-7, 2015. (BIB) (PDF) (DOI)
• , Estimation and Verification of Hybrid Heart Models for Personalised Medical and Wearable Devices. Technical Report CS-RR-15-05, Department of Computer Science, University of Oxford, 2015. (BIB) (PDF) (DOI)
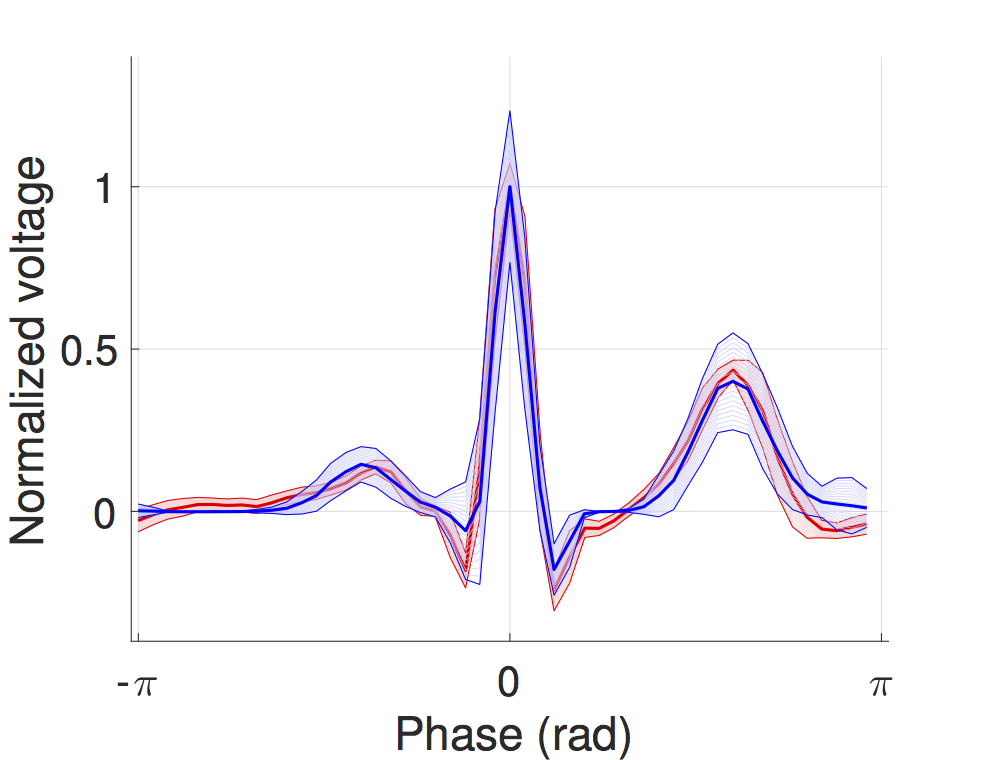
Probabilistic timed modelling of cardiac dynamics and personalization from ECG
We developed a timed automata translation of the IDHP (Integrated Dual-chamber Heart and Pacer) model by Lian et al. Timed components realize the conduction delays between functional components of the heart and action potential propagation between nodes is implemented through synchronisation between the involved components. In this way, the model can be easily extended with other accessory conduction pathways in order to reproduce specific heart conditions.
The IDHP model can be also parametrised from patient electrocardiogram (ECG) in order to reproduce the specific physiological characteristics of the individual. For this purpose, we extended the model in order to generate synthetic ECG signals that reflect the heart events occurring at simulation time. Starting from raw ECG recordings, our method automatically finds model parameters such that the synthetic signal best mimics the input ECG, by minimising the statistical distance between the two. The resulting parameters correspond to probabilistic delays in order to reflect variability of ECG features.
The IDHP heart model can be downloaded from the tool page, while personalization from ECG data is implemented in the HeartVerify tool.
• , Closed-loop quantitative verification of rate-adaptive pacemakers. In ACM Transactions on Cyber-Physical Systems, 2 (4), 2018. (BIB) (PDF) (DOI)
• , Estimation and verification of hybrid heart models for personalised medical and wearable devices. In Computational Methods in Systems Biology (CMSB 2015), pages 3-7, 2015. (BIB) (PDF) (DOI)
• , Estimation and Verification of Hybrid Heart Models for Personalised Medical and Wearable Devices. Technical Report CS-RR-15-05, Department of Computer Science, University of Oxford, 2015. (BIB) (PDF) (DOI)
SMT-based synthesis of gene regulatory networks
Unraveling the structure and the logic of gene regulation is crucial to understand how organisms develop and so is the derivation of predictive models able to reproduce experimental observations and explain unknown biological interactions.
This research centers on the synthesis of biological programs, with specific focus on Boolean gene regulatory networks (GRN). We developed methods based on the idea of synthesis by sketching, which enables explicit modeling of hypotheses and unknown information, specified as e.g. choices or uninterpreted functions. Through the formalization as an SMT problem, the method can automatically and efficiently resolve the unknown information in order to obtain a model that is consistent with observations.
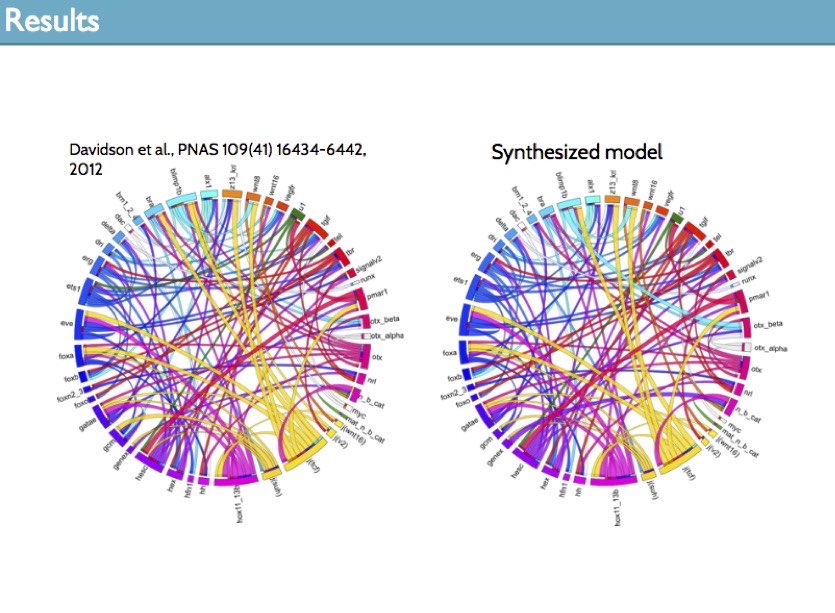
We applied this approach to synthesize the first GRN model of the sea urchin embryo (an important model organism in biology) that precisely and fully reproduce available temporal and spatial expression data and the effects of perturbation experiments. The data we used is the result of 30+ years of research in the Davidson lab at Caltech.
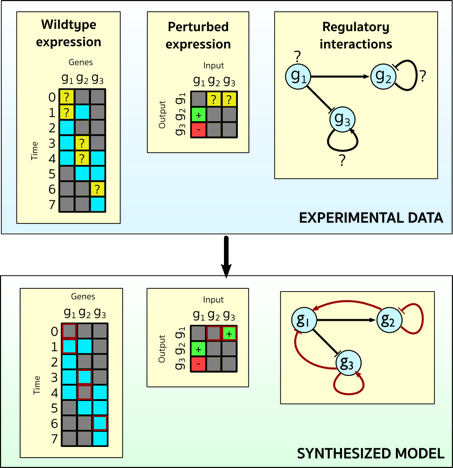
• , Analyzing and Synthesizing Genomic Logic Functions. In Computer Aided Verification (CAV 2014), pages 343-357, 2014. (BIB) (PDF) (DOI)
Network analysis for bioaccumulation and bioremediation in contaminated food webs
In this project we develop computational methods and models to study pollution dynamics in ecological networks and to shed light on three key questions: How persistent organic pollutants (e.g. PCBs that bind to the fat tissue) are transferred in food webs through feeding connections? What are the species that play role in the pollutant distribution? How to synthesize effective bioremediation strategies mediated by pollutant-degrading bacteria?
We present a computational framework to 1) reconstruct bioaccumulation networks from (partial) data; 2) identify key species in contamination dynamics through a new network index based on sensitivity analysis; and 3) analyze the multiscale effects of microbial bioremediation on species-level contamination by integrating metabolic networks of biodegrading bacteria and bioaccumulation networks.
We consider the case of PCBs bioaccumulation in the Adriatic food web and aerobic PCBs bioremediation by a strain of P. putida (see the Tools page to download the models).
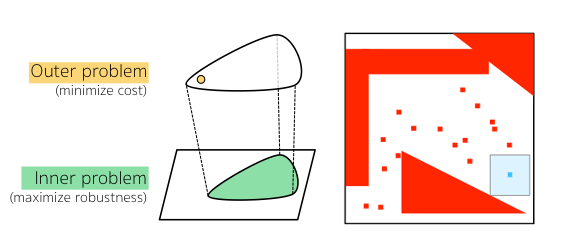
Parameter synthesis for pacemaker design optimization
Verification is useful in establishing key correctness properties of cardiac pacemakers, but has limitations, in that it is not clear how to redesign the model if it fails to satisfy a given property. Instead, parameter synthesis aims to automatically find optimal values of parameters to guarantee that a given property is satisfied.
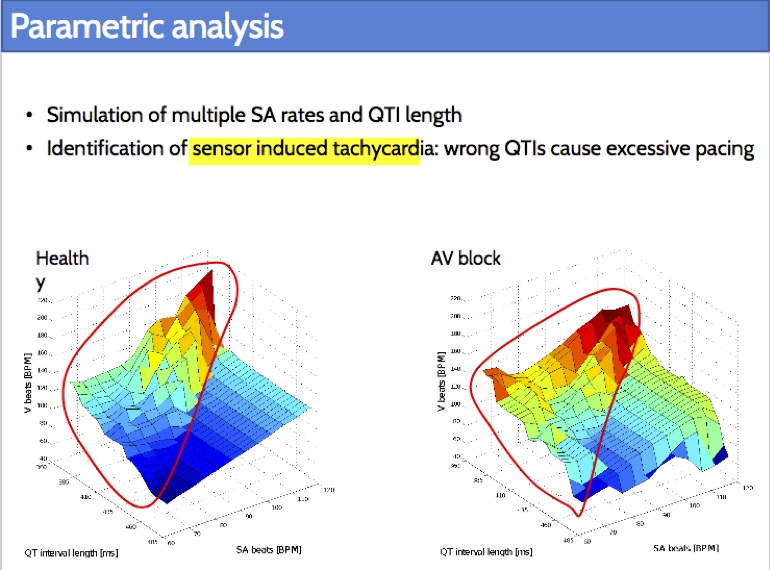
In this project, we develop methods to synthesize pacemaker parameters that are safe, robust and, at the same time, able to optimise a given quantitative requirement such as energy consumption or clinical indicators. We solve this problem by combining symbolic methods (SMT solving) for ruling out parameters that violate heart safety (red areas in the figure) with evolutionary strategies for optimising the quantitative requirement.

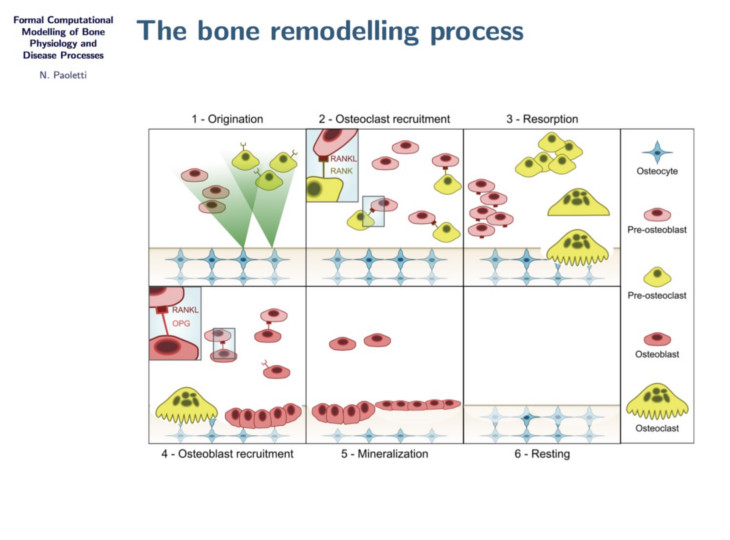
Formal analysis of bone remodelling
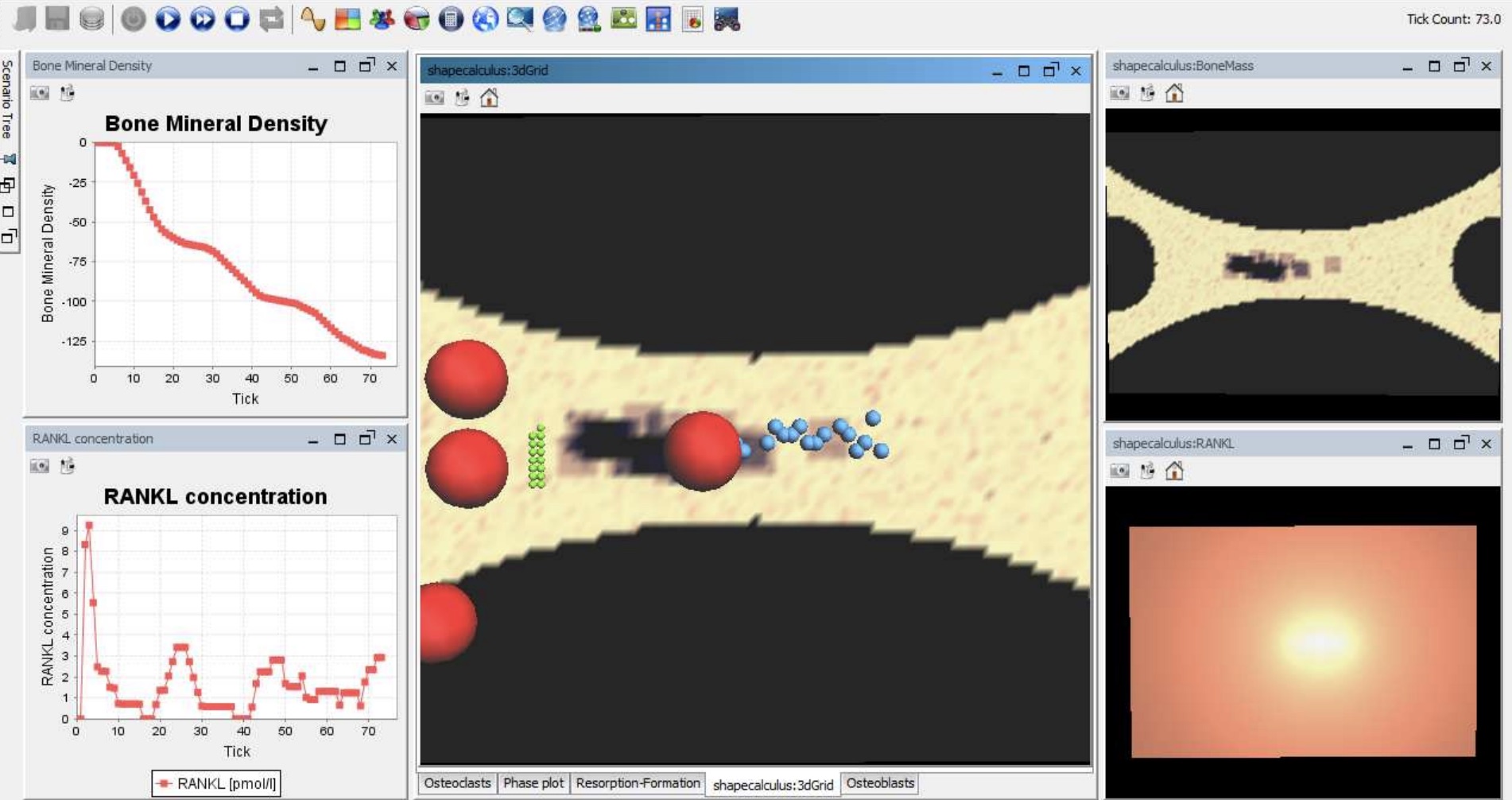
In this project we study bone remodelling, i.e., the biological process of bone renewal, through the use of computational methods and formal analysis techniques. Bone remodelling is a paradigm for many homeostatic processes in the human body and consists of two highly coordinated phases: resorption of old bone by cells called osteoclasts, and formation of new bone by osteoblasts. Specifically, we aim to understand how diseases caused by the weakening of the bone tissue arise as the resulting process of multiscale effects linking the molecular signalling level (RANK/RANKL/OPG pathway) and the cellular level.
To address crucial spatial aspects such as cell localisation and molecular gradients, we developed a modelling framework based on spatial process algebras and a stochastic agent-based semantics, realised in the Repast Simphony tool. This resulted in the first agent-based model and tool for bone remodelling, see also the Tools page.
In addition, we explored probabilistic model checking methods to precisely assess the probability of a given bone disease to occur, and also hybrid approximations to tame the non-linear dynamics of the system.
• , Modelling osteomyelitis. In BMC bioinformatics, 13 (14): S12, 2012. (BIB) (PDF) (DOI)
• , Multilevel computational modeling and quantitative analysis of bone remodeling. In IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB), 9 (5): 1366-1378, 2012. (BIB) (PDF) (DOI)
• , Multiple Verification in Complex Biological Systems: The Bone Remodelling Case Study. In Trans. Computational Systems Biology, 14: 53-76, 2012. (BIB) (PDF) (DOI)
• , A combined process algebraic and stochastic approach to bone remodeling. In Electronic Notes in Theoretical Computer Science, 277: 41-52, 2011. (BIB) (PDF) (DOI)
• , Osteoporosis: a multiscale modeling viewpoint. In Computational Methods in Systems Biology (CMSB 2011), pages 183-193, 2011. (BIB) (PDF) (DOI)
• , Multiple verification in computational modeling of bone pathologies. In Computational Models for Cell Processes (CompMod 2011), pages 82-96, 2011. (BIB) (PDF) (DOI)